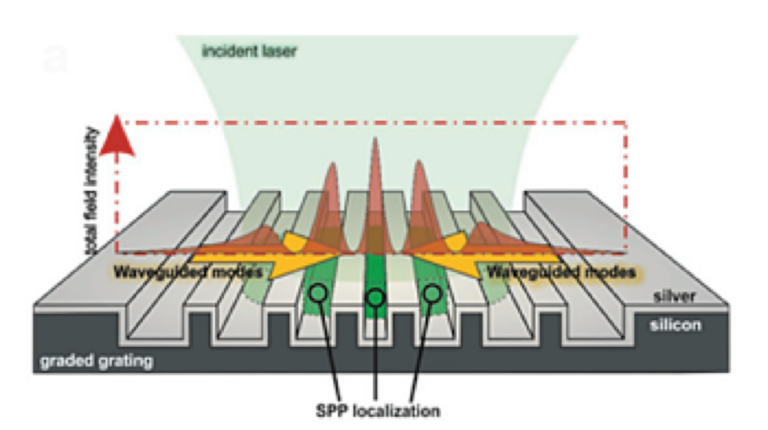
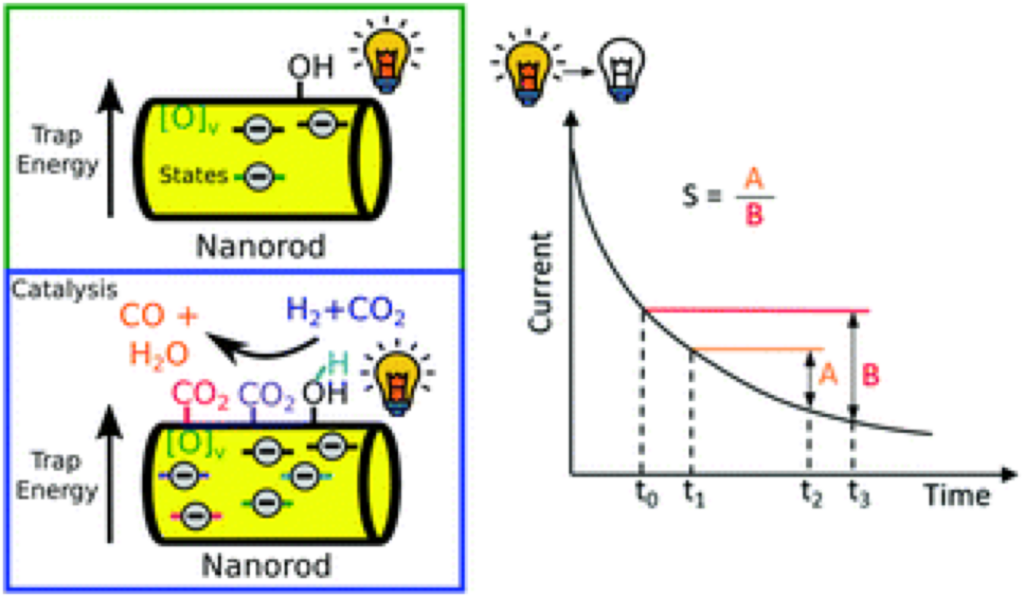
We are a research group led by Prof. Nazir P. Kherani in the Electrical and Computer Engineering Department at the University of Toronto. The Advanced Photonics-Photovoltaics & Devices (AP2D) Laboratory supports and fosters interdisciplinary education and research in the areas of photovoltaics and the allied fields of photonics and electronics.
The AP2D Group at the University of Toronto is actively carrying out innovative research and technological advancements. The AP2D Group works closely with industry and research partners, helping develop innovative technologies and conducting fundamental and applied research. AP2D and its affiliated laboratories consist of state-of-the-art thin film and nanostructured materials and device synthesis and diagnostic facilities.
Research advances has been made by securing federal grants such as NSERC alliance, NSERC RTI, ORF-RE, Canadian Institutes of Health Research, Mitacs award and CMC.
Congratulations to Dr. Katelyn Dixon for starting a post-doctorate position at Quantum Valley Ideas Lab, Dr. Sidra Farid for starting a sessional faculty position at the McMaster University and Mahdi Safari for accepting an industry position respectively.
Outreach event was organized in coordination with Let’s Talk Science to high school students to cultivate interest in sensing and light-based sciences.
Moein Shayegannia, with Mitacs support, participated in a 16-week Lab2Market program spearheaded by Dalhousie University that instilled skills required to assess the commercial opportunity from lab to market.
A PoC Sensing Symposium was held on 18th November, 2021 at the University of Toronto by the ORF-RE Sensing Team that brought together leaders from industry, clinicians and academia to share their knowledge and explore synergies.